




- Escrito por Lilian Russo
- Imprimir
- Seja o primeiro a comentar!
Aprovado registro de produto de terapia gênica
 Produto é indicado para o tratamento da atrofia muscular espinhal.A Anvisa publicou, nesta segunda-feira (17/8), o registro do produto de terapia gênica Zolgensma® (onasemnogeno abeparvoveque), da empresa Novartis Biociências S.A. Confira aqui a publicação.
Produto é indicado para o tratamento da atrofia muscular espinhal.A Anvisa publicou, nesta segunda-feira (17/8), o registro do produto de terapia gênica Zolgensma® (onasemnogeno abeparvoveque), da empresa Novartis Biociências S.A. Confira aqui a publicação.
O Zolgensma é um produto de terapia gênica usado para o tratamento da atrofia muscular espinhal (AME), uma doença rara e grave, causada pela alteração do gene que codifica a proteína SMN
(survival motor neuron), molécula necessária para a sobrevivência do neurônio motor, responsável pelo controle do movimento muscular. A AME causa fraqueza, hipotonia, atrofia e paralisia muscular progressiva e possui incidência mundial aproximada de 1:10.000 nascidos vivos, dos quais cerca de 45% a 60% dos casos são AME do tipo 1, forma mais grave da doença, sendo a principal causa de mortalidade infantil decorrente de uma doença monogenética.
O Zolgensma obteve o registro na Anvisa para o tratamento de pacientes pediátricos diagnosticados com AME do tipo 1, com até 2 anos de idade, com mutações bialélicas no gene de SMN1 ou até três cópias de outro gene conhecido como SMN2.
Engenharia genética
Desenvolvido por engenharia genética, é um produto de terapia avançada, do tipo terapia gênica, composto por um vetor viral que carrega uma cópia funcional do gene humano responsável pela produção da proteína SMN, capaz de restaurar a função do neurônio motor no organismo dos pacientes.
Os estudos realizados até o momento com o Zolgensma demonstraram que uma aplicação única do produto pode melhorar a sobrevivência dos pacientes, reduzir a necessidade de ventilação permanente para respirar e alcançar marcos de desenvolvimento motores. Os efeitos colaterais do produto são considerados controláveis, sendo que o mais comum observado nos estudos foi o aumento das enzimas hepáticas, resolvido após tratamento com corticosteroides.
A Anvisa avaliou, portanto, que os benefícios do Zolgensma são superiores aos seus riscos e pode ser registrado no Brasil. No entanto, por ser uma terapia gênica inovadora, foi aprovado um registro de caráter excepcional. Isso significa que estudos adicionais devem ser realizados pela empresa para a confirmação de sua eficácia e segurança em longo prazo.
Termo de compromisso
Nesse sentido, foi assinado um Termo de Compromisso em que a Novartis se responsabiliza por enviar anualmente dados dos estudos em andamento à Anvisa e ainda assegurar a realização de estudos complementares de acompanhamento de pacientes brasileiros, de forma a acompanhar o perfil de segurança e de eficácia do produto no país em longo prazo, na perspectiva da avaliação da manutenção do balanço benefício x risco positivo, como atestado no registro.
As informações técnicas sobre o Zolgensma e as condições impostas ao detentor de registro no Termo de Compromisso serão informadas publicamente, junto com prazos e datas para sua execução, por meio do portal da Agência.
Regularização
A Anvisa, após desenvolver o marco regulatório para o registro de produto de terapia avançada no Brasil com a publicação da Resolução da Diretoria Colegiada (RDC) 338/2020, criou um ambiente regulatório seguro para a aprovação de produtos dessa natureza no país.
O processo de registro do Zolgensma envolveu a análise de:
- Comprovação de segurança, por meio de dados de experimentos pré-clínicos.
- Comprovação de segurança e eficácia, por meio de dados de estudos clínicos.
- Comprovação de produção com requisitos de qualidade e boas práticas de fabricação na indústria produtora nos Estados Unidos.
- Estudos de estabilidade e mecanismos de distribuição do produto no Brasil, bem como processos controlados de importação.
- Avaliações das estratégias para orientações e precauções de cuidados especiais ao paciente.
- Estratégias de monitoramento e gerenciamento de risco após a administração do produto aos pacientes no Brasil.
O processo de registro envolveu também a responsabilização da empresa pelo treinamento dos profissionais responsáveis pela infusão do produto, bem como pela qualificação prévia dos estabelecimentos hospitalares brasileiros para o correto armazenamento e controle do produto antes de sua administração.
Vale ressaltar que a Anvisa também integrou ao processo avaliativo do registro os processos de Certificação de Boas Práticas de Fabricação (CBPF) dos produtores do componente ativo e do produto final, concluindo, por meio de análise documental, que o processo de fabricação do Zolgensma nos Estados Unidos demonstra ter qualidade consistente e controlada.
O processo de avaliação técnica do novo produto foi efetivado por quatro especialistas da Agência, com a participação de três consultores ad hoc da Rede Nacional de Especialistas em Terapias Avançadas e da Câmara Técnica (CAT) de Terapias Avançadas da Anvisa, formada por cientistas das principais instituições de pesquisa do Brasil.
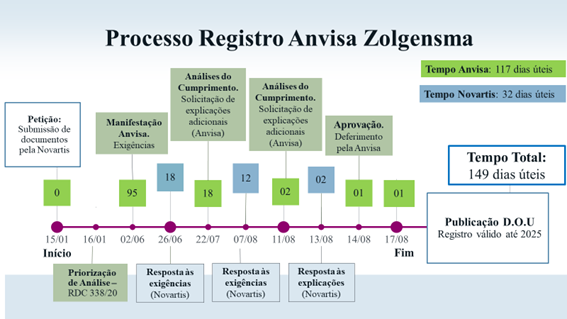
Prazo
Foram 149 dias úteis de avaliação, desde a submissão dos documentos pela Novartis à Anvisa até a publicação do deferimento final no Diário Oficial da União, considerando os prazos de análise da equipe da Agência (117 dias) e das respostas ao cumprimento das exigências por parte da empresa (32 dias). Para garantir a efetiva regularização do Zolgensma no Brasil, houve inúmeras reuniões, discussões técnicas e acordos entre a Anvisa e representantes nacionais e internacionais.
Com a publicação do registro a Agência cumpre a legislação nacional, que prevê um prazo de análise pela instituição de 120 dias para produtos priorizados.
Considerando a complexidade deste produto de tecnologia avançada, os prazos aplicados na Anvisa foram compatíveis com os das principais agências reguladoras do mundo:
Prazo total FDA (Food and Drugs Administration/EUA) – aproximadamente 167 dias úteis (submissão 01/10/2018 – aprovação 31/05/2019).
Fonte: https://www.fda.gov/media/127961/download
Prazo total EMA (European Medicines Agency) – aproximadamente 406 dias úteis (submissão 08/10/2018 – aprovação 18/05/2020).
Fonte: https://www.ema.europa.eu/en/documents/assessment-report/zolgensma-epar-public-assessment-report_en.pdf
O produto também foi avaliado, em relação à biossegurança de organismos geneticamente modificados, pela Comissão Nacional Técnica de Biossegurança (CTNBio), que se manifestou igualmente favorável à sua aprovação no país.
Saiba mais:
O que é produto de terapia avançada?
É uma categoria especial de medicamentos novos que compreende os produtos de terapia celular avançada, os produtos de engenharia tecidual e os produtos de terapia gênica.
E o produto de terapia gênica?
Do ponto de vista regulatório, conceitua-se como um produto de origem biológica cujo componente ativo contenha ou consista em ácido nucleico recombinante, podendo ter o objetivo de regular, reparar, substituir, adicionar ou deletar uma sequência genética e/ou modificar a expressão de um gene, com vistas a um resultado terapêutico, preventivo ou de diagnóstico.